Our paper about estimating alternative polyadenylation (APA) from scRNA-seq has been accepted by Nucleic Acids Research.
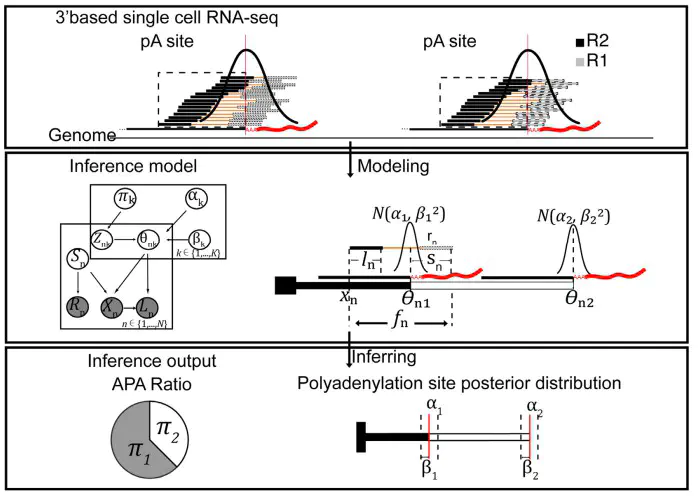
Alternative polyadenylation increases transcript diversities at the 3’end, regulating biological processes including cell differentiation, embryonic development and cancer progression. Here, we present a Bayesian method SCAPE, which enables de novo identification and quantification of polyadenylation (pA) sites at single-cell level by utilizing insert size information. We demonstrated its accuracy and robustness and identified 31 558 sites from 36 mouse organs, 43.8%(13 807) of which were novel. We illustrated that APA isoforms were associated with miRNAs binding and regulated in tissue-, cell typeand tumor-specific manners where no difference was found at gene expression level, providing an extra layer of information for cell clustering. Furthermore, we found genome-wide dynamic changes of APA usage during erythropoiesis and induced pluripotent stem cell (iPSC) differentiation, suggesting APA contributes to the functional flexibility and diversity of single cells. We expect SCAPE to aid the analyses of cellular dynamics and diversities in health and disease.